
Un UDI (Unique Device Identifier) est un code unique au monde pour identifier des dispositifs médicaux. Il est composé de deux parties au moyen de caractères numériques ou alphanumériques: un identifiant de dispositif (DI) unique et un identifiant de production (PI) variable.
GS1 est un organisme accrédité pour l'émission des UDI qui vous aide à se conformer aux réglementations internationales, comme EU MDR IVDR et US FDA, qui exigent des fournisseurs qu'ils attribuent un UDI à leurs produits.

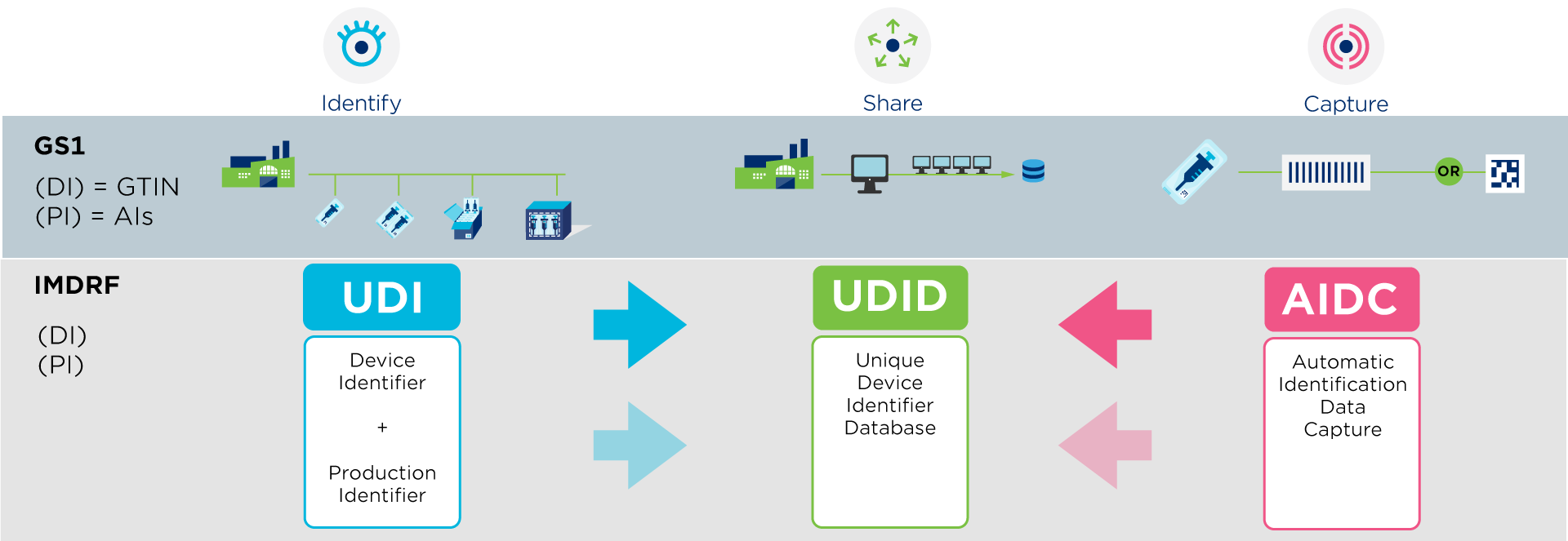
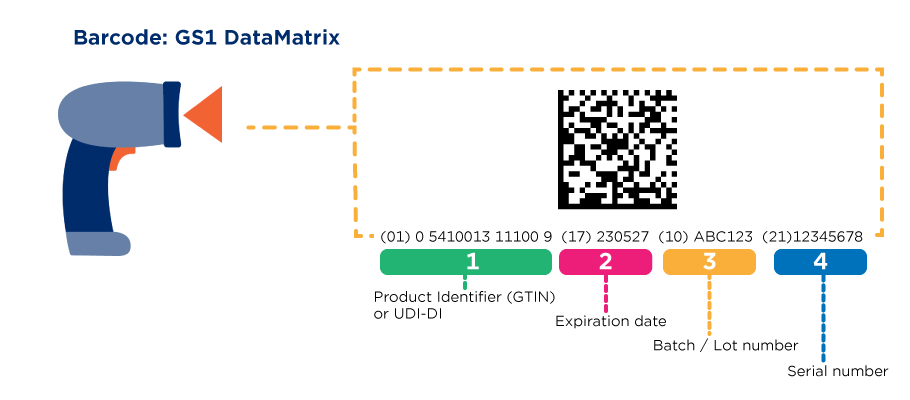
En tant que fournisseur, vous êtes tenu d'enregistrer vos données (=UDI-DI) dans une base de données réglementée organisée par le gouvernement.
Pour l'Amérique (FDA), il s'agit de GUDID et vous pouvez y publier votre UDI-DI à partir de My Product Manager.
Pour l'Europe, c'est EUDAMED. Vous pouvez publier vos données via My Basic UDI-DI Manager. Contactez-nous par e-mail via healthcare@gs1belu.org, si cela vous intéresse.
Découvrez My Basic UDI-DI Manager
